Fully functional bundle: To use the fully functional ImageJ bundle, just download the platform-dependent version of ImageJ bundled with Java file and containing all the tools needed for the package embedded:
JEasyTFM command at the right of the ImageJ menu for using it.

Manual installation: Proceed with the manual installation by downloading the ImageJ software and unziping it.
Alternatively the download of all the previously described files can simply be replaced by downloading the JEasyTFM_files file and unzip it in the plugins/jars folder. Finally one of the platform-dependent library files need to be downloaded into the plugins/jars folder:
|
|||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||||

|
The JEasyTFM tool is divided into two submenus: 
Splitting the JEasyTFM tool into two steps, with on one side a job creation and on the other side, a job launching operation, provides two main advantages. The first one is to offer traceability of all the analysis settings used to generate the different analysis results. In the case of heavily booked dedicated analysis computers, the second one is to rapidly define and protect the analysis settings, which can then quickly be launched later on once the machine is available. When launching the JEasyTFM->Create_job tab, the following window will be displayed: 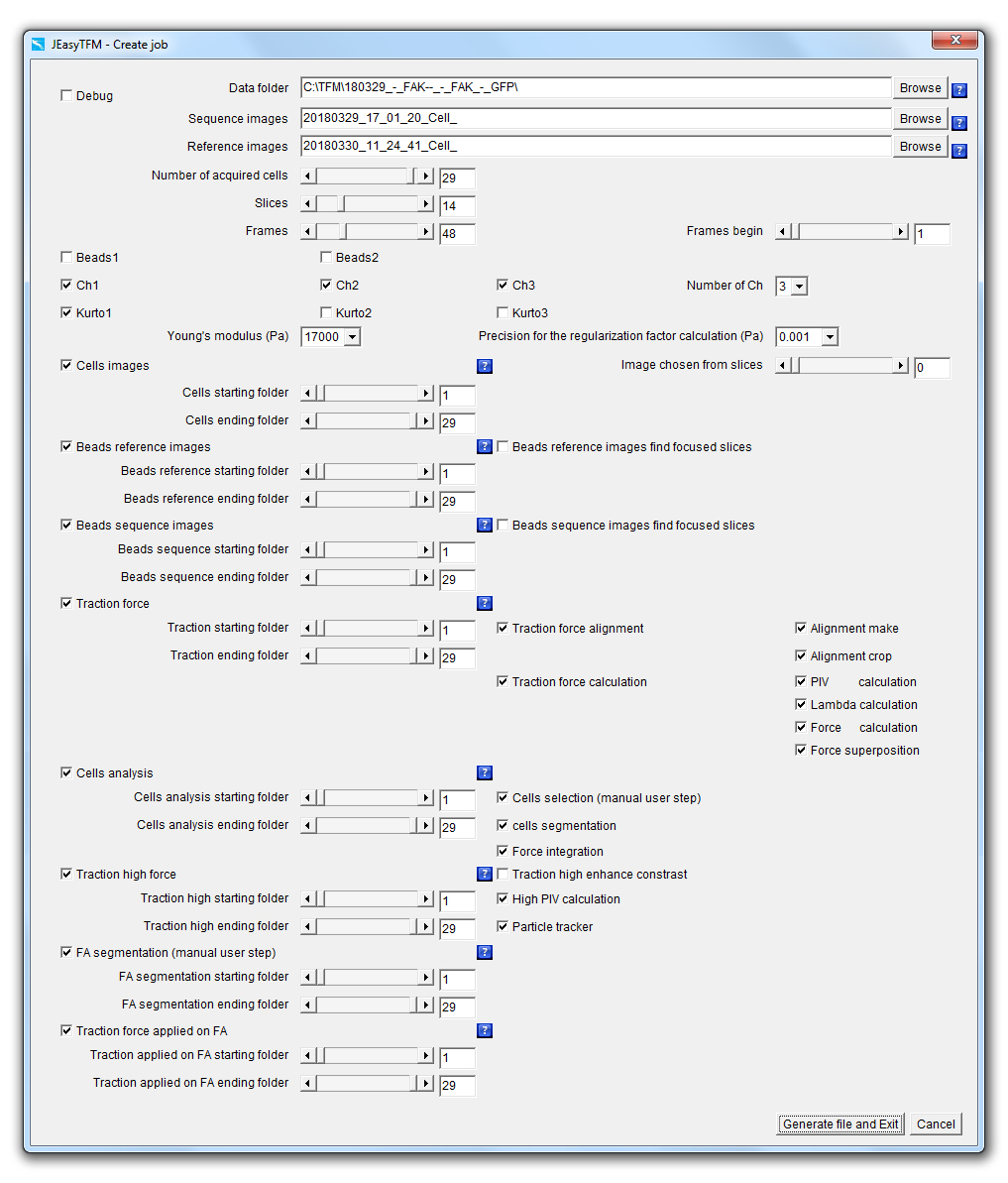
We will now explain each point in details.
|

Debug: When the Debug checkbox is selected, the analysis is performed by launching the different used plugins going through their GUI, often resulting in images being displayed at each stage for the user to visualize the results of the different processes going on. On the contrary, when the Debug checkbox is unselected, all the analyses are performed launching the additional plugins by using some back-doors (i.e. static methods) that have been created to short-cut the plugins GUI resulting in all the images produced in memory and not displayed for a gain of machine time. Data_folder: On clicking on the Data_folder Browse button, a "choose directory" dialog is displayed for the user to choose the directory in which the data to be analyzed are saved.
Upon selection validation of the chosen folder, its path will be updated within the field Data_folder together with the prefixes file name of the acquired images to be analyzed in the File field and the one for the images of the beads at equilibrium (i.e. after the addition of trypsin also called reference images in this document) in the Reference_images field.
Sequence_images: On clicking on the Sequence_images Browse button, a "file open" dialog is displayed for the user to choose an image file to be analyzed.
Upon selection validation of an image file, the file name prefix of the acquired images to be analyzed will be updated within the Sequence_images field.
Reference_images: On clicking on the Reference_images Browse button, a "file open" dialog is displayed for the user to choose a reference images file to be analyzed.
Upon selection validation of the image file, the file name prefix of the acquired images to be analyzed will be updated within the Reference_images field.
The reference images selected in Reference_images have to be previously saved within a folder whose name is the one defined within Data_folder field with the addition of "_-_Reference".
|

Number_of_acquired_cells: Slider defining the number of cells that have been recorded during the acquisitions.
Note that the value specified by the Number_of_acquired_cells slider represents the maximum value for the sliders:
Slices: Slider defining the number of z-slices that have been acquired during the acquisitions. Frames: Slider defining the number of time-frames that have been acquired during the acquisitions. Frames_begin: Slider whose maximum value will be defined by the Frames slider and defining the first time-frames number of the data that will be analyzed.
|

Beads1: The selection of the Beads1 and/or Beads2 checkboxes implies that an experiment of high-resolution TFM has been performed, i.e. with the use of beads with two different colors.
Beads2: The selection of the Beads2 and/or Beads1 checkboxes implies that an experiment of high-resolution TFM has been performed, i.e. with the use of beads with two different colors.
Ch1: The selection of the Ch1 and/or Ch2 and/or Ch3 checkboxes implies that more than one channel has been acquired for imaging the cells.
Ch2: The selection of the Ch1 and/or Ch2 and/or Ch3 checkboxes implies that more than one channel has been acquired for imaging the cells.
Ch3: The selection of the Ch1 and/or Ch2 and/or Ch3 checkboxes implies that more than one channel has been acquired for imaging the cells.
Number of Ch: Popup menu defining the number of channels that have been acquired.
Kurto1: The Kurto1 checkbox will apply a best Kurtosis selection algorithm to the images of the Ch1 in the case it is selected and the "Extended Depth Field"1 plugin in the case it is unselected.
Kurto2: The Kurto2 checkbox will apply a best Kurtosis selection algorithm to the images of the Ch2 in the case it is selected and the "Extended Depth Field"1 plugin in the case it is unselected.
Kurto3: The Kurto3 checkbox will apply a best Kurtosis selection algorithm to the images of the Ch3 in the case it is selected and the "Extended Depth Field"1 plugin in the case it is unselected.
Young's_modulus_Pa: Numerical field defining the Young's modulus in Pascal units of the support used for cell plating. Precision_for_the_Regularization_factor_calculation: Popup menu defining the precision for the calculation of the regularization or Lagrange parameter λ value that is used to convert the measured beads displacements to the forces applied from the cells onto their support.
(1) Forster, B., Van De Ville, D., Berent, J., Sage, D. & Unser, M. Complex Wavelets for Extended Depth-of-Field: A New Method for the Fusion of Multichannel Microscopy Images. Microsc. Res. Tech. 65, 33-42 (2004).
|

Cells_images: The checkbox defines whether cell images should be extracted from all the Slices for all the defined Frames and for the cells numbers defined between the Cells_starting_folder and Cells_ending_folder.
Cells_starting_folder: Defines the first cell number or folder for which cell images should be extracted from all the Slices and for all the defined Frames.
Cells_ending_folder: Defines the last cell number or folder for which cell images should be extracted from all the Slices and for all the defined Frames.
Image_chosen_from_slices: The automatic selection of the best focused image may be performed by putting the value 0 in this slider. In this case, a Kurtosis algorithm will be applied on the Slices images in order to extract the one giving the highest value and this for all the defined Frames. On the other hand, manual inspection of the z-slices can be performed and the slice number of the selected best focused image can be defined in the Image_chosen_from_slices slider by choosing which cell image number between 1 and the number indicated in Slices should be extracted from all the Slices for all the defined Frames. |

Beads_Reference_images: The checkbox defines whether images corresponding to the best focused image of the beads extracted within the z-series acquired with the support at equilibrium (i.e. after the cells have been detached from the surface) should be extracted from all the Slices.
Beads_reference_starting_folder: Defines the first cell number or folder for which images corresponding to the best focus of the beads within the images acquired with the support at equilibrium (i.e. after the cells have been detached from the surface) should be extracted from all the Slices.
Beads_reference_ending_folder: Defines the last cell number or folder for which images corresponding to the best focus of the beads within the images acquired with the support at equilibrium (i.e. after the cells have been detached from the surface) should be extracted from all the Slices.
Beads_Reference_images_find_focused_slices: The checkbox defines whether the "Find focused slices"2 plugin should be applied prior to applying the "Extended Depth Field"1 plugins on the images of the beads with the support at equilibrium from all the Slices in order to extract the best focused image.
(2) Tseng, Q. Find Focused Slices. ImageJ plugin available at: https://sites.google.com/site/qingzongtseng/find-focus
(1) Forster, B., Van De Ville, D., Berent, J., Sage, D. & Unser, M. Complex Wavelets for Extended Depth-of-Field: A New Method for the Fusion of Multichannel Microscopy Images. Microsc. Res. Tech. 65, 33–42 (2004). |

Beads_Sequence_images: The checkbox defines whether images corresponding to the best focus of the beads extracted within the images through the acquired time-lapse should be extracted from all the Slices for all the defined Frames.
Beads_sequence_starting_folder: Defines the first cell number or folder for which images corresponding to the best focus of the beads extracted within the images through the acquired time-lapse should be extracted from all the Slices for all the defined Frames.
Beads_sequence_ending_folder: Defines the last cell number or folder for which images corresponding to the best focus of the beads extracted within the images through the acquired time-lapse should be extracted from all the Slices for all the defined Frames.
Beads_Sequence_images_find_focused_slices: The checkbox defines whether the "Find focused slices"2 plugin should be applied prior to applying the "Extended Depth Field"1 plugin on the images of the beads through the acquired time-lapse, defining the Frames from all the Slices for all the defined Frames to extract the best focused image.
(2) Tseng, Q. Find Focused Slices. ImageJ plugin available at: https://sites.google.com/site/qingzongtseng/find-focus
(1) Forster, B., Van De Ville, D., Berent, J., Sage, D. & Unser, M. Complex Wavelets for Extended Depth-of-Field: A New Method for the Fusion of Multichannel Microscopy Images. Microsc. Res. Tech. 65, 33–42 (2004). |

Traction_force: The checkbox defines whether the whole or part of the force calculation processes should be launched.
This is the main checkbox for the analysis, which means that if it is not selected, none of the following analysis processes:
If the checkboxes Beads1 and/or Beads2 is/are activated, the launched analysis will then be applied to all the selected beads colors. Traction_starting_folder: Defines the first cell number or folder for which the whole or part of the force calculation processes should be calculated.
Traction_ending_folder: Defines the last cell number or folder for which the whole or part of the force calculation processes should be calculated.
Traction_force_alignment: The checkbox defines whether the whole or part of the images alignment processes should be calculated.
This means that if the checkbox Traction_force_alignment is not selected none of the following images alignment processes: will be launched whether or not they are selected. If the checkboxes Beads1 and/or Beads2 is/are activated, the launched analysis will then be applied to all the selected beads colors. Alignment_make: The checkbox defines whether an alignment algorithm should be launched on the images.
Alignment_crop: The checkbox defines whether the algorithm that will get rid of the image borders generated by the Alignment_make algorithm should be launched on the images.
Traction_force_calculation: The checkbox defines whether the whole or part of the images forces calculation processes should be launched.
This means that if the checkbox Traction_force_calculation is not selected none of the following force calculation processes: will be launched whether or not they are selected. If the checkboxes Beads1 and/or Beads2 is/are activated, the launched analysis will then be applied to all the selected beads colors. PIV_calculation: The checkbox defines whether a PIV (Particle Image Velocimetry) algorithm should be launched on the images of the aligned and cropped beads in order to generate beads displacement maps.
Lambda_calculation: The checkbox defines whether the regularization or Lagrange parameter λ values should be calculated.
Force_calculation: The checkbox defines whether a Fourier Transform Traction Cytometry (FTTC) algorithm should be launched on the beads displacement maps previously generated with the PIV_calculation feature in order to create force maps.
Force_superposition: The checkbox defines whether the force maps previously generated with the Force_calculation feature should be superimposed with the images of the cells that have been previously generated with the Cells_images feature.
|

Cells_analysis: The checkbox defines whether the whole or part of the cells segmentation and force integration processes should be launched. This is the main checkbox for the analysis, which means that if it is not selected none of the following analysis processes:
will be launched whether or not they are selected.
If the checkboxes Beads1 and/or Beads2 is/are activated, the launched analysis will then be applied to all the selected beads colors. Cells_analysis_starting_folder: Defines the first cell number or folder for which the whole or part of the cells segmentation and force integration processes should be launched.
Cells_analysis_ending_folder: Defines the last cell number or folder for which the whole or part of the cells segmentation and force integration processes should be launched.
Cells_selection (manual user step): The checkbox defines whether a cells segmentation algorithm with some manual user steps should be launched on the images of the cells that have been previously aligned and cropped with the Alignment_make and Alignment_crop algorithms and pre-filtered with the Cells_segmentation algorithm.
Cells_segmentation: The checkbox defines whether a cells segmentation algorithm should be launched on the images of the cells that have been previously aligned and cropped with the Alignment_make and Alignment_crop algorithms.
Force_integration: The checkbox defines whether a whole cells force integration algorithm should be launched on the traction force images using the cells segmentation ROI (Region Of Interest) data that have been obtained either automatically through a Cells_selection (manual user step) algorithm or manually and the result together with the ROI drawing superimposed with the images of the cells that have been previously generated with the Cells_images feature.
|

Traction_high_force: The checkbox defines whether the whole or part of the beads positions tracking processes should be launched.
This is the main checkbox for the analysis, which means that if it is not selected none of the following analysis processes: will be launched whether or not they are selected. If the checkboxes Beads1 and/or Beads2 is/are activated, the launched analysis will then be applied to all the selected beads colors. Traction_high_starting_folder: Defines the first cell number or folder for which the whole or part of the beads positions tracking processes should be calculated.
Traction_high_ending_folder: Defines the last cell number or folder for which the whole or part of the beads positions tracking processes should be calculated.
Traction_high_enhance_contrast: The checkbox defines whether a Subtract_Background... followed by an Enhance_Contrast... filter should be applied on the images in order to enhance their contrast before applying the High_PIV_calculation regression.
High_PIV_calculation: The checkbox defines whether a PIV (Particle Image Velocimetry) algorithm should be launched on the images of the aligned and cropped beads in order to generate beads displacement maps.
Particle_tracker: The checkbox defines whether a particle tracking algorithm should be launched on the beads displacement maps previously generated with the High_PIV_calculation feature in order to track the beads displacements between images that have been previously generated with the Beads_Sequence_images.
|

FA_segmentation (manual user step): The checkbox defines whether a FA_segmentation (manual user step) algorithm should be launched on fluorescent images of the cells that have been previously aligned and cropped with the Alignment_make and Alignment_crop algorithms.
FA_segmentation_starting_folder: Defines the first cell number or folder for which the FA_segmentation (manual user step) algorithm should be applied.
FA_segmentation_ending_folder: Defines the last cell number or folder for which the FA_segmentation (manual user step) algorithm should be applied.
|

Traction_force_applied_on_FA: The checkbox defines whether a Traction_force_applied_on_FA7-8 algorithm should be launched which will calculate the forces applied on the focal adhesion positions that had previously been determined by the FA_segmentation algorithm using the beads displacements data obtained by the Particle_tracker algorithm.
Traction_force_applied_on_FA_starting_folder: Defines the first cell number or folder for which the Traction_force_applied_on_FA algorithm should be applied.
Traction_force_applied_on_FA_ending_folder: Defines the last cell number or folder for which the Traction_force_applied_on_FA algorithm should be applied.
(7) Schwarz, U. S., Balaban, N. Q., Riveline, D. Bershadsky, A., Geiger, B., Safran, S. A.
Calculation of forces at focal adhesions from elastic substrate data: the effect of localized force and the need for regularization, Biophys. J. 83, 1380-1394 (2002) (8) Sabass, B., Gardel, M. L., Waterman, C. and Schwarz, U. S. high-resolution traction force microscopy based on experimental and computational advances, Biophys. J., 94,207-220, (2008) |

Generate_file_and_Exit: The button will validate the selections within the JEasyTFM>Create_job window, save the data of the analysis configuration job into a batch file (JEasy.txt) within the ImageJ plugins folder and close the JEasyTFM>Create_job window. Cancel: The button will cancel the selections within the JEasyTFM>Create_job window, without saving the data of the analysis configuration job into a batch file (JEasy.txt) and close the JEasyTFM>Create_job window. |

 for all the acquired cells will be extracted.
The extracted images will be named
BeadsReference.tif
In the case of the activation of the checkbox
Beads1
the images will be named
BeadsReference_1.tif
and in the case of the activation of the checkbox
Beads2
the images will be named
BeadsReference_2.tif
The images will be saved in a folder named Cell z; z corresponding to the given cell number.")



















